Sampling Rare Conformational Transitions with a Quantum Computer
Abstract
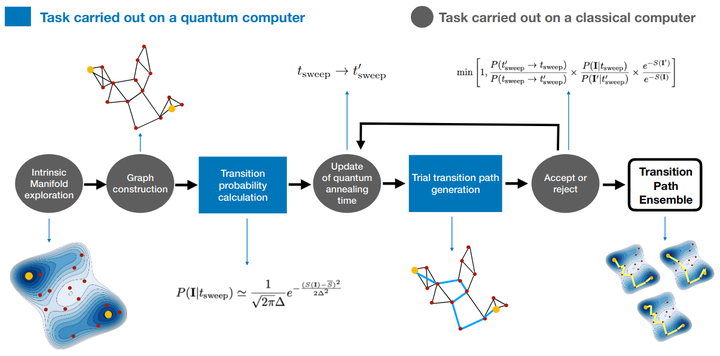
Spontaneous structural rearrangements play a central role in the organization and function of complex biomolecular systems. In principle, physics-based computer simulations like Molecular Dynamics (MD) enable us to investigate these thermally activated processes with an atomic level of resolution. However, rare conformational transitions are intrinsically hard to investigate with MD, because an exponentially large fraction of computational resources must be invested to simulate thermal fluctuations in metastable states. Path sampling methods like Transition Path Sampling hold the great promise of focusing the available computational power on sampling the rare stochastic transition between metastable states. In these approaches, one of the outstanding limitations is to generate paths that visit significantly different regions of the conformational space at a low computational cost. To overcome these problems we introduce a rigorous approach that integrates a machine learning algorithm and MD simulations implemented on a classical computer with adiabatic quantum computing. First, using functional integral methods, we derive a rigorous low-resolution representation of the system’s dynamics, based on a small set of molecular configurations generated with machine learning. Then, a quantum annealing machine is employed to explore the transition path ensemble of this low-resolution theory, without introducing un-physical biasing forces to steer the system’s dynamics. Using the D-Wave quantum computer, we validate our scheme by simulating a benchmark conformational transition in a state-of-the-art atomistic description. We show that the quantum computing step generates uncorrelated trajectories, thus facilitating the sampling of the transition region in configuration space. Our results provide a new paradigm for MD simulations to integrate machine learning and quantum computing.